
“Scientists have gotten pretty good at sequencing DNA, determining the order of the bases in an individual’s DNA, but we’re not good at understanding what we see,” Chris Tyler-Smith, a geneticist at the UK’s Wellcome Trust Sanger Institute, told Singularity Hub.
Even as whole-genome sequencing has become much cheaper, genetic research has continued to focus on the exome, or the tiny fraction of human DNA that codes the proteins that make up our physical structures.
But according to a recent study published in Science and conducted by Tyler-Smith and others, the sources of cancer most often lie in the rest of the genome, once referred to as junk DNA.
Cancer is, in other words, a very important needle in a very large haystack. The study also helpfully provides a roadmap to the spots in the genetic haystack that are most likely to give rise to cancer.
By comparing data from two open-access genetic databases, ENCODE and the 1000 Genomes project, Tyler-Smith and his colleagues were able to identify parts of the non-coding genome that rarely show mutations. Such stability suggests that evolution does not smile on such mutations — in other words, they’re harmful.
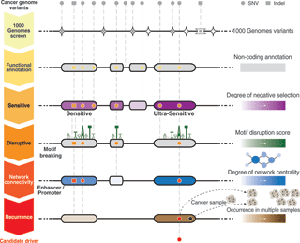 With that schema in hand, the researchers then looked at cancer genomes from open-access sources and found that mutations in the stable locations occurred repeatedly, making a statistical case that they’re relevant to cancer.
With that schema in hand, the researchers then looked at cancer genomes from open-access sources and found that mutations in the stable locations occurred repeatedly, making a statistical case that they’re relevant to cancer.
“In cancers, lots of mutations occur and most are irrelevant. But there’s a small number that cancer researchers call the ‘drivers,’ meaning they’re responsible for making that cancer grow or expand. The ‘passenger’ mutations occur at random, but for the important ‘driver’ mutations, you see the same mutation or a mutation in the same gene again and again,” Tyler-Smith explained.
Pointing to these likely driver mutations, the authors hope to help focus future cancer drug research on agents that affect those mutations and eventually to trigger cancer treatments targeting a wider range of genetic signatures.
The National Institutes of Health’s Cancer Genome Atlas, which shares the goal of identifying specific cancer-causing mutations, also indicates that this kind of map-making is needed before personalized cancer treatments can benefit more than the small fraction of patients they reach today.
“There are many steps needed to translate genomic data to patient care, including determining which genomic changes are truly responsible for each cancer and identifying or developing therapies to correct the impact of those changes,” the NIH says on the project’s website.
 Certainly sequencing cancers has become more common. But given the abundance of “irrelevant” mutations, the sequences don’t often give clinicians actionable information. (Singularity Hub also recently covered a search engine for genes found in cancer cells, where oncologists can look to see if there is any research suggesting that a given gene is more or less responsive to a particular drug.)
Certainly sequencing cancers has become more common. But given the abundance of “irrelevant” mutations, the sequences don’t often give clinicians actionable information. (Singularity Hub also recently covered a search engine for genes found in cancer cells, where oncologists can look to see if there is any research suggesting that a given gene is more or less responsive to a particular drug.)
“DNA tells us a lot about where the broken parts are, and that’s critical for targeting our fixes. For example, if your car is making a rumbling in the front, you need to know if it’s in the engine or the transmission. And that’s sort of what we don’t know currently. We know what these cells are broken in some way, but we’re not always able to tell where the break is so that we can target it with a therapy,” Neil Hayes, a U.N.C. Chapel Hill oncologist, explains in an NIH video.
The Science study gets researchers a lot closer.
It may also have secondary effects on the quickly changing field of genetics. It verifies big data approaches, first of all. The study authors said similar statistic analyses could also help find genetic bases for diseases other than cancer. The findings also add to the growing body of research that says what was once called “junk” DNA is anything but.
Images: Juan Gaertner via Shutterstock.com; Khurana, Tyler-Smith and Gerstein via Science; Suse Broyde via Wikimedia Commons